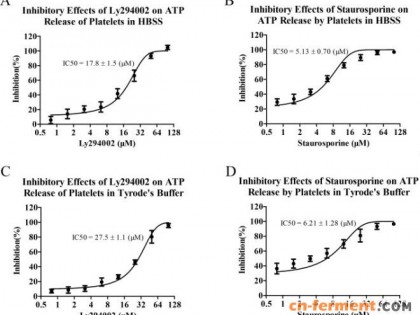
Staurosporine (星形孢菌素) 简称STS,是一种 ATP 竞争性和非选择性的蛋白激酶抑制剂,对 PKC、PKA、c-Fgr、Phosphorylase kinase 和 TAOK2 均有抑制活性,可作为凋亡诱导剂,常用于细胞凋亡机制以及癌症相关研究。
作用机制
Staurosporine 可能通过激活胱天蛋白酶-3等凋亡相关蛋白来触发细胞凋亡程序诱导细胞凋亡。在较低浓度下,Staurosporine 还能根据细胞类型的不同,诱导特定的细胞周期效应,如阻止细胞在G1期或G2/M期的进程。
此外,Staurosporine 还具备细胞毒性、松弛平滑肌和调节 eNOS 基因表达等多种生物学作用,这些作用共同构成了其复杂的药理效应网络(如图1)。因此,其主要作用机制可分为以下几点:
蛋白激酶抑制:Staurosporine是一种广谱的蛋白激酶抑制剂,能够抑制多种蛋白激酶的活性,如PKC、PKA等。它通过占据激酶上的ATP结合位点,阻止ATP与激酶的相互作用,从而阻断激酶的催化功能。
诱导细胞凋亡:在特定浓度下,Staurosporine能够诱导多种细胞类型的凋亡。这一作用机制涉及激活凋亡相关蛋白(如胱天蛋白酶-3)和调控细胞周期等过程。
神经保护:在较低浓度下,Staurosporine还具有一定的神经保护功能,能够促进神经突起的生长
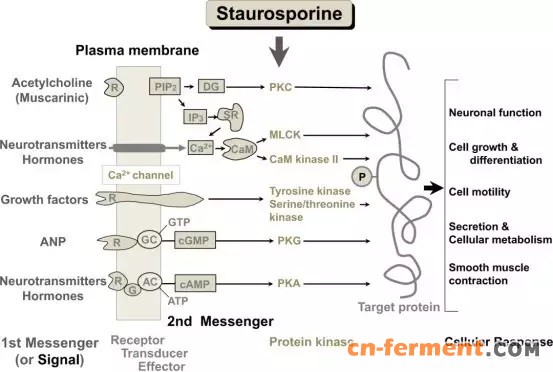
▲Staurosporine的信号转导和抑制模型[1]
适用研究方向
Staurosporine 的药理活性在于其强效的蛋白激酶抑制能力。通过竞争性地结合激酶上的ATP结合位点,Staurosporine能够有效抑制包括PKC(蛋白激酶C)、PKA(蛋白激酶A)、PKG(蛋白激酶G)在内的多种激酶,其中对PKC-α的抑制活性尤为显著,比PKC-δ和PKC-ζ高出数百至数千倍。这种强大的抑制能力使得Staurosporine在诱导细胞凋亡、阻断细胞周期等方面展现出卓越的药理效果。
抗癌研究
Staurosporine 是一种非常有效但非特异性的蛋白激酶抑制剂,特别是酪氨酸激酶,并且它对癌细胞具有非常强的细胞毒性作用。已知超过 90 种酪氨酸激酶对恶性转化和肿瘤血管生成至关重要。酪氨酸激酶抑制剂 (TKI) 可以靶向受体激酶和细胞质激酶,可以通过控制癌细胞中激酶的激活来改善癌症预后。为了研究癌细胞凋亡的潜在机制,Staurosporine被用作乳腺癌细胞系的凋亡刺激物(如图2)。
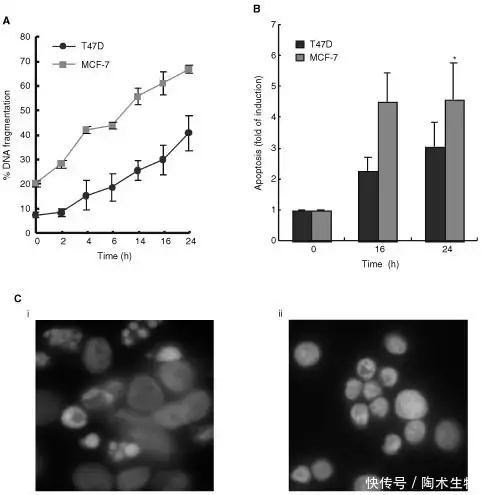
▲Staurosporine诱导 T47D 和 MCF-7 人乳腺癌细胞凋亡[2]
细胞信号传导与细胞周期调控
Staurosporine 作为一种非特异性的蛋白激酶抑制剂,通过抑制多种蛋白激酶的活性,影响细胞信号传导和细胞周期调控。研究人员利用 Staurosporine 作为工具,探讨了不同蛋白激酶在细胞生长、分化、凋亡等过程中的作用。
研究表明,Staurosporine 可导致胃癌细胞停滞在 G2/M 期,这可能是抑制细胞增殖并导致这些细胞凋亡的机制之一。Staurosporine 对 G2/M 期细胞的影响可能归因于 p21WAF1 基因的上调(如下图)。
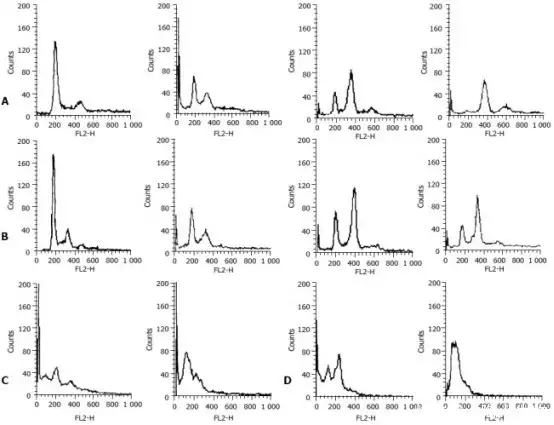
▲Staurosporine诱导胃癌细胞系 MGC803 和 SGC7901 细胞 G2/M 期停滞[3]
神经科学研究
在神经科学研究领域,Staurosporine 因其神经保护功能而受到关注。有研究表明己酮可可碱对Staurosporine诱导的 PC12 细胞神经突生长过程有积极影响[4];同样,在较低浓度下,Staurosporine能够促进神经突起的生长,为神经退行性疾病的治疗提供了新的思路。
相关研究证实 1nM Staurosporine可有效防止 NGF 剥夺 4 天后约 50% 的交感神经神经元死亡,对应浓度的神经元均保持完整的神经突、细胞体以及正常的核形态(如图4);因此 Staurosporine 可以作为神经突生长的诱导剂,在神经退行性疾病相关研究中发挥着十分重要的作用。
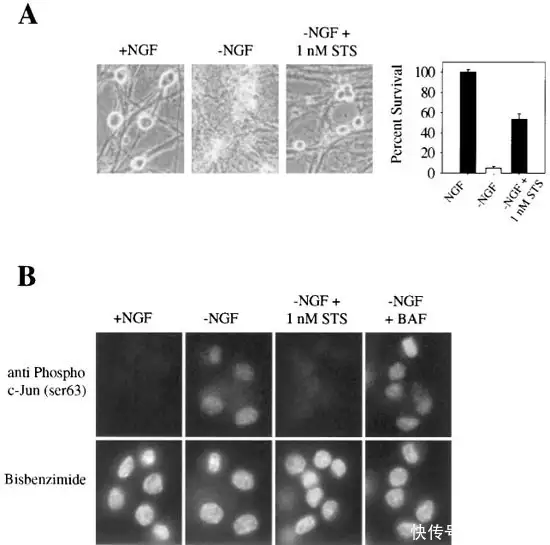
▲极低浓度的Staurosporine(1 nM) 可防止交感神经元死亡[5]
药物开发
鉴于 Staurosporine 的多种生物活性,研究人员还在探索其作为药物开发的可能性。通过化学合成和基因工程等技术手段,研发具有更高生物活性和更低毒性Staurosporine衍生物,以期在抗癌、神经保护等领域展现出更好的应用前景。
Staurosporine 的类似物 UCN-01 和 CGP 41251 均已被证明能在体外有效阻止几种人源性肿瘤细胞系的生长。如 UCN-01 可阻断 CRC-SC 的生长,增强伊立替康在体外和体内 CRC-SC 衍生模型中的活性(如下图)。
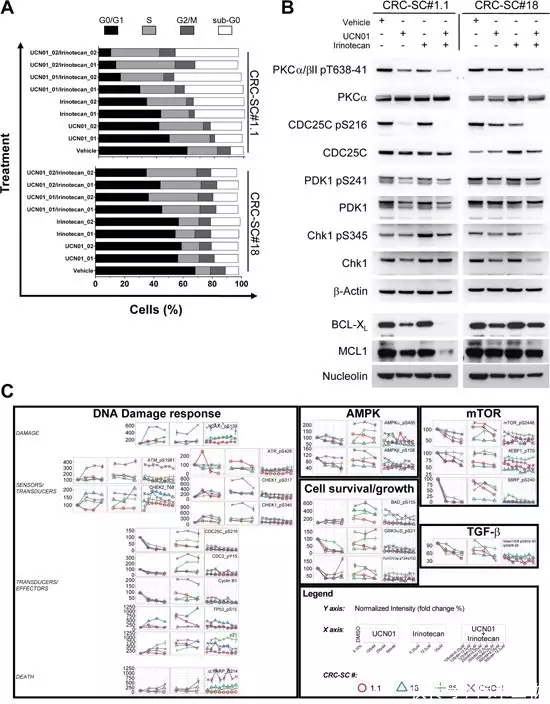
▲Staurosporine的类似物UCN-01 与伊立替康的组合通过靶向 DNA 损伤通路阻断 CRC-SC 复制[6]
Staurosporine 作为一种强效的ATP竞争性激酶抑制剂,自1977年被发现以来,其独特的药理活性和广泛的应用潜力一直吸引着科研界的广泛关注。这种从链霉菌staurosporines中分离出来的天然产物,以其对多种激酶的高亲和力抑制作用而闻名,尤其在抗癌治疗领域展现出巨大潜力。尽管其临床应用因缺乏特异性而受到限制,但作为一种研究工具,Staurosporine在探索抗癌机制、筛选抗癌药物等方面发挥着重要作用。
同样,Staurosporine能够影响细胞周期的不同阶段,为研究细胞增殖、分化等生物学过程提供了有力的工具。除了抗癌和细胞周期研究外,Staurosporine还展现出抗真菌、抗高血压、抗菌、抗病毒、抗炎等多种生物活性。因此,这种多样的生物活性为未来基于Staurosporine所开展的药物设计与改造,神经保护(如阿尔茨海默病、帕金森病)、心血管疾病(心肌保护、抗心律失常)等研究提供了新的思路以及广阔的开发空间。